





O Brasil possui um quantitativo de medicamentos similares elevado, e isso faz parte da cultura e do histórico do País. Assim, um profissional de assuntos regulatórios precisa conhecer melhor o processo de registro de medicamentos similares.
De acordo com a professora do ICTQ – Instituto de Pesquisa e Pós-Graduação para Mercado Farmacêutico, dra. Luciana Colli, medicamento similar é aquele que contém os mesmos princípios ativos, concentração, forma farmacêutica, via de administração, posologia e indicação terapêutica (preventiva ou diagnóstica) do medicamento de referência registrado no órgão federal responsável pela vigilância sanitária. No entanto, eles podem diferir somente em características relativas ao tamanho e forma do produto, prazo de validade, embalagem, rotulagem, excipientes e veículos, devendo sempre ser identificados por nome comercial ou marca.
Arcabouço legal de registro de medicamentos similares:
- Lei 6360/76: estabelece às regras de registro de produtos vinculado a vigilância sanitária. Criou Lei de Medicamentos - similar que podia ou não apresentar marca.
- Decreto 8077/13 (antigo: 79094/77): regulamenta a Lei 6360/76;
- Lei 9.782/99: Criação da Agência Nacional de Vigilância Sanitária (Anvisa);
- Lei 9787/99: altera a Lei 6360/76, criando o medicamento genérico, dispõe sobre a utilização de nomes genéricos em produtos farmacêuticos e dá outras providências;
- Decreto 3181/99: regulamenta a Lei 9787/99.
Por meio da Resolução 3.916/98, a Política Nacional de Medicamentos tem como objetivo:
- estimular a concorrência e a variedade de oferta no mercado de medicamentos;
- promover o uso de medicamentos genéricos;
- melhorar a qualidade de todos os medicamentos;
- reduzir os preços; e
- facilitar o acesso da população aos tratamentos.
“Como consequência, houve o estímulo ao desenvolvimento de medicamentos genéricos e adequação técnica do medicamento similar. A resolução que regula o registro de medicamento similar é RDC 200, publicada em 28 de outubro de 2017”, comenta Luciana.
Diferença entre medicamento similar, inovador e referência
Referência: medicamento inovador registrado no órgão federal responsável pela vigilância sanitária e comercializado no País, cuja eficácia, segurança e qualidade foram comprovadas cientificamente junto ao órgão federal competente, por ocasião do registro (Lei 9.787, de 10 de fevereiro de 1999).
Inovador: medicamento comercializado no mercado nacional composto por, pelo menos, um fármaco ativo, sendo que esse fármaco deve ter sido objeto de patente, mesmo já extinta, por parte da empresa responsável por seu desenvolvimento e introdução no mercado do país de origem, ou o primeiro medicamento a descrever um novo mecanismo de ação, ou aquele definido pela Anvisa que tenha comprovado eficácia, segurança e qualidade.
Similar: aquele que contém os mesmos princípios ativos, concentração, forma farmacêutica, via de administração, posologia e indicação terapêutica. Ele é equivalente ao medicamento registrado no órgão federal responsável pela vigilância sanitária, podendo diferir somente em características relativas ao tamanho e forma do produto, prazo de validade, embalagem, rotulagem, excipientes e veículos, devendo sempre ser identificado por nome comercial ou marca.
“A principal diferença entre eles é que o similar é identificado por uma marca/nome comercial. O genérico é identificado pela Denominação Comum Brasileira (DCB) ou Denominação Comum Internacional (DCI) e pela faixa amarela com o G (genérico), não podendo adotar nome de marca”, explica Luciana.
Procedimentos de registro de um medicamento similar
De acordo com a RDC 200/17 existem os procedimentos antecedentes ao registro, o registro em si e, posteriormente, as medidas pós-registro, que ocorrem após a publicação do registro na Anvisa.
Como medida antecedente ao registro, é preciso verificar a existência do fármaco em questão em algum medicamento de referência, para a conferência da indicação, concentração e forma farmacêutica. Caso não exista um medicamento de referência com o fármaco desejado é necessário apontar um de referência, com o seguinte procedimento: cadastrar a empresa, produto, princípio ativo, forma farmacêutica, concentração e comprovante de comercialização/distribuição no Brasil.
De acordo com a RDC 200/17, Art. 20, não serão admitidos para fins de registro como medicamento genérico ou similar:
I - produtos biológicos, imunoterápicos, derivados do plasma e sangue humano;
II - medicamentos fitoterápicos;
III - medicamentos específicos;
IV - medicamentos dinamizados;
V - medicamentos de notificação simplificada;
VI - antissépticos de uso hospitalar;
VII - produtos com fins diagnósticos e contrastes radiológicos;
VIII- radiofármacos;
IX - gases medicinais; e
X - outras classes de medicamentos que venham a possuir legislação específica para seu registro.
No processo de registro de medicamentos similares existem os documentos administrativos e os técnicos. Para os documentos administrativos há:
- Formulários de Petição;
- Via de Pagamento da Guia de Recolhimento da União (GRU);
- Licença de Funcionamento vigente;
- Certificado de Responsabilidade Técnica vigente;
- Autorização de Funcionamento;
- Autorização Especial de Funcionamento; e
- Certificado de Boas Práticas de Fabricação (CBPF).
Como documentos técnicos:
- Relatório de Produção;
- Ordem de produção (OP);
- Relatório de Controle de qualidade: laudo, especificações e metodologia;
- Validação;
- Drug Master File (DMF); e
- Especificação de material de embalagem.
Relatório técnico, por forma farmacêutica:
- Tamanho(s) do(s) lote(s) industrial (ais) a ser (em) produzido(s), incluindo mínimo e máximo;
- Descrição de todas as etapas do processo de produção contemplando os equipamentos utilizados, detalhamento do desenho, do princípio de funcionamento e da capacidade máxima individual;
- Quando houver diluente: cópia de dossiês completos de produção e controle de qualidade do diluente; e
- Descrição dos critérios de identificação do lote industrial.
Ordem de produção:
- Cópia de três dossiês completos de produção e controle de qualidade de três lotes pilotos notificados, com inclusão da ordem de produção (cálculos explicativos, fichas de limpeza e pesagem), contemplando também a etapa de embalagem primária; e
- Para medicamentos com três ou mais concentrações diferentes e formulações proporcionais, apresentar os dossiês de produção e controle de qualidade da menor e da maior concentração.
Relatório de controle de qualidade do produto acabado:
- Especificações e métodos analíticos, a referência bibliográfica farmacopeica;
- Caso a metodologia não seja farmacopeica, ou farmacopeica com adaptações, apresentar validação;
- Validação de testes para determinação do teor, determinação de impurezas e produtos de degradação, teste de dissolução e teste de identificação; e
- Laudo de análise.
Relatório de controle de qualidade de excipientes:
- Citar a referência bibliográfica adotada no controle de qualidade de todos os excipientes;
- No caso de excipiente não descrito em compêndios oficiais, apresentar as especificações e os métodos de análise adotados; e
- Apresentar cópia do laudo analítico de controle de qualidade do(s) excipiente(s), realizado pela empresa.
Relatório de controle de qualidade do fármaco:
- Apresentar as especificações e métodos analíticos, a referência bibliográfica farmacopeica;
- Caso a metodologia não seja farmacopeica, apresentar validação;
- Oferecer validação para determinação do teor e de impurezas e produtos de degradação;
- Laudo; e
- Drug Master File (DMF).
Papel timbrado da empresa produtora do ativo, incluindo:
- Dados gerais da empresa fabricante com o endereço completo do local de fabricação do fármaco;
- Rota de síntese, com a descrição das moléculas intermediárias, seus nomes químicos e solventes utilizados;
- Descrição das especificações e métodos analíticos adotados pelo fabricante do fármaco e cópia do laudo analítico do controle de qualidade fornecido pelo mesmo;
- Quantificação e limites dos principais contaminantes, de acordo com a rota de síntese do fármaco;
- Quantificação dos solventes residuais;
- No caso de quiralidade, apresentar os dados sobre os teores dos estereoisômeros, quando a proporção desses estereoisômeros possa comprometer a eficácia e a segurança do medicamento;
- No caso de polimorfismo, apresentar metodologia analítica adotada e resultados dos testes de determinação dos prováveis polimorfos do fármaco;
- Validação dos métodos analíticos empregados, quando não seguirem metodologia farmacopeica; e
- Cópias dos laudos analíticos de controle de qualidade, fornecido pelo fabricante do fármaco.
Especificação de material de embalagem
É o documento que descreve detalhadamente o material de embalagem primária utilizado. “Deve-se enviar especificações e métodos analíticos utilizados no controle de qualidade da embalagem primária do medicamento e dos acessórios. O acessório dosador para administração do medicamento deve estar em quantidades adequadas considerando sua posologia”, lembra Luciana.
Na regulamentação - RDC 305/02, RDC 68/03 e RDC 208/18 – para as substâncias derivadas de ruminantes:
- Certificado de boas práticas de fabricação do fabricante;
- Laudo analítico de CQ;
- Certificado Veterinário Internacional (CVI); ou
- Certificado de Conformidade - Farmacopéia Européia.
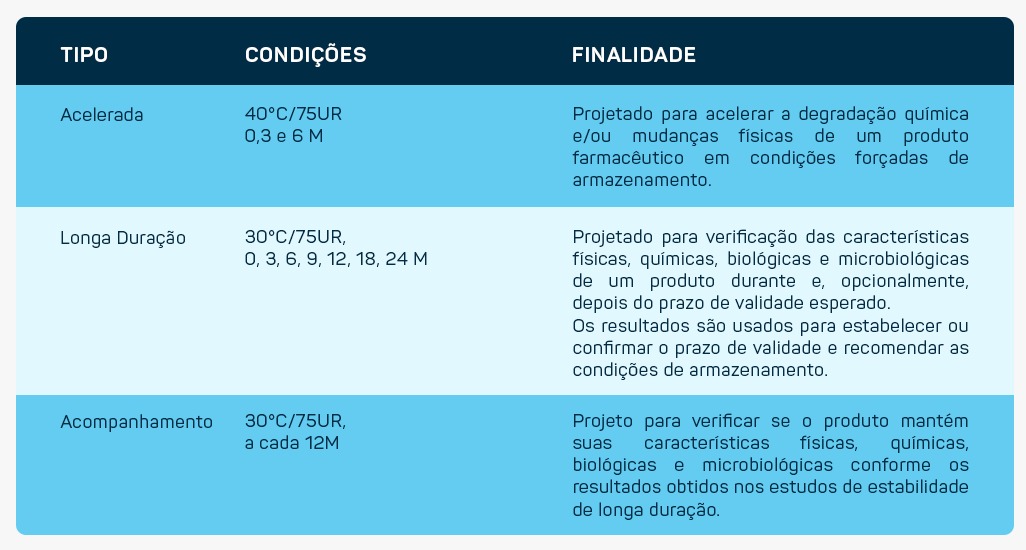
Estudo de estabilidade
A regulamentação – RE 01/2005 – é o Guia para a Realização de Estudo de Estabilidade. Os principais fatores de influência na estabilidade são processo, excipientes, características físico-químicas do ativo, material de embalagem, temperatura, umidade e luz.
A determinação de prazo de validade inclui o período provisório de 24 meses, acelerado de 6 meses, acompanhado do estudo de longa duração, ou estudo de longa duração de 12 meses.;
Com o foco ná Anvisa, são necessários estudo de fotoestabilidade, estudo de reconstituição (informação da bula) e produtos de degradação.
Bula
Os itens obrigatórios para a bula do paciente e do profissional de saúde são:
- Nome comercial ou marca;
- DCB;
- Apresentações comercializadas, citando: forma farmacêutica, concentração, quantidade total de peso, volume líquido ou unidades farmacotécnicas;
- Quantidade total de dosadores, quando aplicável;
-Via de administração;
- USO ADULTO ACIMA DE... e/ou USO PEDIÁTRICO ACIMA DE ....
- Composição do ativo e excipientes usando a DCB;
- Produtos líquidos e em gotas: equivalência de gotas para cada mililitro (gotas/mL) e massa por gota (mg/mL);
- Número de registro com 9 dígitos;
- Farmacêutico responsável e número de inscrição no CRF;
- Razão social e endereço do detentor do registro no Brasil;
- CNPJ do detentor do registro;
- Para os medicamentos fabricados e/ou embalados por empresas diferentes da detentora do registro, informar a razão social da empresa fabricante e local de fabricação do produto, citando a cidade e o estado, precedido pelas expressões, conforme o caso: "Fabricado por:" e "Embalado por:";
- Para os produtos importados, discriminar o local de fabricação do medicamento, citando a cidade, o estado e país, e incluir as seguintes expressões, conforme o caso: "Importado por:"; "Fabricado por:"; "Embalado por:";
- Telefone do Serviço de Atendimento ao Consumidor (SAC);
- Frases obrigatórias, quando for o caso: "Uso restrito a hospitais", "Uso profissional", "Venda sob prescrição médica", "Dispensação sob prescrição médica" (para laboratórios oficiais).
- Incluir, exceto nos textos de bula a serem submetidos eletronicamente à Anvisa, uma das seguintes frases, conforme o caso, em negrito:
"Esta bula foi aprovada pela Anvisa em (dia/mês/ano)" (informando a data de publicação da bula no Bulário Eletrônico)”;
"Esta bula foi atualizada conforme Bula Padrão aprovada pela Anvisa em (dia/mês/ano)" (informando a data de publicação da respectiva Bula Padrão no Bulário Eletrônico com a qual a bula foi harmonizada e/ou atualizada)”;
-Incluir símbolo da reciclagem de papel;
Equivalência Farmacêutica
A resolução RDC 31/10 aborda a equivalência farmacêutica e perfil de dissolução, incluindo:
- Fabricação de três lotes-piloto;
- Estudo de estabilidade acelerada e longa duração com três lotes;
- Estudo de equivalência farmacêutica com um lote; e
- O mesmo lote da equivalência farmacêutica é submetido ao estudo de bioequivalência (Biolote).
Biodisponibilidade
“O teste de biodisponibilidade é muito importante para o medicamento similar, pois ele permite que o mesmo seja intercambiável, tal como o genérico”, ressalta Luciana.
A biodisponibilidade relaciona-se à quantidade absorvida e a velocidade do processo de absorção do fármaco liberado a partir da forma farmacêutica administrada. Quando dois medicamentos de ação sistêmica apresentam a mesma biodisponibilidade no organismo, sua eficácia clínica é considerada comparável.
Isso indica a velocidade e a extensão de absorção (quantidade) de um princípio ativo em uma forma de dosagem, a partir de sua curva concentração versus o tempo (curva PK) na circulação sistêmica ou sua excreção na urina.
A biodisponibilidade absoluta de um medicamento administrado sob a forma de solução injetável intravenosa é total (100%), uma vez que toda a dose administrada está disponível para exercer o efeito.
Biodisponibilidade relativa (BDR)
Trata-se de uma comparação (razão) entre duas biodisponibilidades do mesmo principio ativo, podendo ser a partir de vias de administração diferentes ou não, usando parâmetros farmacocinéticos específicos (Cmax e ASC0-t).
Bioequivalência (BE)
“Consiste na demonstração de equivalência farmacêutica entre produtos apresentados sob a mesma forma farmacêutica, contendo idêntica composição qualitativa e quantitativa de princípio(s) ativo(s), e que tenham comparável biodisponibilidade (in vivo), quando estudados sob um mesmo desenho experimental”, comenta Luciana.
Na etapa clínica elabora-se o protocolo clínico para a administração dos medicamentos teste e referência. Posteriormente é realizada a coleta das amostras do material biológico (sangue, plasma, soro, urina etc.) dos voluntários sadios previamente recrutados. Esta etapa deve ser realizada sob Boas Práticas Clínicas (BPC).
A etapa analítica é quando os fármacos são quantificados nas amostras coletadas durante a etapa clínica por um método bioanalítico validado. A etapa deve ser realizada sob Boas Práticas de Laboratório (BPL).
De acordo com a professora, na etapa estatística, após quantificação das concentrações do fármaco, aplica-se um método estatístico apropriado para analisar os dados e verificar os parâmetros da análise de bioequivalência. Ao final desta etapa, emite-se um relatório integrado das três etapas, em que consta o parecer com o resultado final da análise.
Bioisenções
De acordo com a RDC 37/11, os estudos de bioequivalência são dispensados para os seguintes tipos de medicamentos:
I - soluções aquosas (parenterais, orais, otológicas, oftálmicas e as administradas como inalatórios orais ou sprays nasais, com ou sem dispositivo) que contenham o mesmo fármaco, na mesma concentração em relação ao medicamento de referência (equivalentes farmacêuticos) e excipientes de mesma função que aqueles presentes no medicamento comparador;
II - pós para reconstituição que resultem em soluções aquosas orais ou parenterais, desde que cumpram os requisitos descritos no inciso I;
III - gases;
IV - soluções oleosas parenterais que contenham o mesmo fármaco, na mesma concentração em relação ao medicamento de referência (equivalentes farmacêuticos) e qualitativamente o mesmo veículo oleoso presente no medicamento de referência, em concentrações compatíveis com a função pretendida;
V - medicamentos de uso oral que contenham fármacos destinados à ação local no trato gastrintestinal descritos na Lista 3 - Fármacos de ação local no trato gastrintestinal que não necessitam de estudos de biodisponibilidade relativa e bioequivalência (acessível no portal da Anvisa); e
VI - medicamentos de aplicação tópica, não destinados a efeitos sistêmicos, que contenham o mesmo fármaco, na mesma concentração em relação ao medicamento de referência (equivalentes farmacêuticos) e excipientes de mesma função que aqueles presentes.
